Band structure#
PyProcar goes beyond the conventional plain band structure to plot the projected bands that carry even more information. The projected bands are color coded in an informative manner to portray fine details.
1. Plain band structure#
This is the most basic type of band structure. No projection information is contained here. In order to use the plain mode one sets mode='plain'. elimit sets the energy window limits. outcar specifies the OUTCAR file. For Abinit calculations, abinit_output is used instead. color lets the user use any color available in the matplotlib package. If an output file is not present one can set fermi manually. One may save the plot using the savefig tag, for example, savefig='figure.png' with a desired image file format. This applies to all other band structure plotting functions in PyProcar as well. For Elk, setting file=’PROCAR’ and outcar=’OUTCAR’ is not necessary. As long as the path to elk.in is defined or PyProcar is executed in the directory of calculation, PyProcar will work of the files needed for the bandsplot.
Usage:
pyprocar.bandsplot(code='vasp',dirname='bands',elimit=[-2,2],mode='plain',color='blue')
PyProcar is capable of labeling the
One may manually label the knames and kticks corresponds to the labels and the number of grid points between the high symmetry points in the
Usage:
pyprocar.bandsplot(code='vasp',dirname='bands',elimit=[-2,2],mode='plain',color='blue',kticks=[0,39,79,119,159],knames=['G','X','M','G','R'])
2. Spin projection#
For collinear spin polarized and non-collinear spin calculations of DFT codes, PyProcar is able to plot the bands considering spin density (magnitude), spin magnetization and spin channels separately.
For non-collinear spin calculations, spins=[0] plots the spin density (magnitude) and spins=[1,2,3]
corresponds to spins oriented in spin='st'
plots the spin texture perpendicular in the plane (mode=`parametric'. cmap refers to the matplotlib color map used for the parametric plotting
and can be modified by using the same color maps used in matplotlib. cmap='seismic' is recommended for parametric spin band structure plots.
For colinear spin calculations setting spins=[0] plots the spin density (magnitude) and spins=[1] plots the spin magnetization.
Spin channels can also be plot separately (see below).
If spin-up and spin-down bands are to be plot separately (for colinear calculations), there are two methods one can follow.
Usage:
pyprocar.bandsplot(code='vasp',dirname='bands',mode='plain', spins=[0]) # plots the spin up channel
pyprocar.bandsplot(code='vasp',dirname='bands',mode='plain', spins=[1]) # plots the spin down channel
These methods can be used for both plain and parametric modes. If a comparison of spin up and spin down bands is required on the same plot, the following syntax can be used.
Usage:
pyprocar.bandsplot(code='vasp',dirname='bands',mode='plain', spins=[0,1]) # plots both the spin up and spin down channels
Note:
Currently, Elk only supports spin colinear plotting. Non colinear spin plotting will be implemented in the future.
3. Atom projection#
The projection of atoms onto bands can provide information such as which atoms contribute to the electronic states near the Fermi level. PyProcar counts each row of ions in the PROCAR file, starting from zero. In an example of a five atom SrVO:math:_3, the indexes of atoms for Sr, V and the three O atoms would be 0,1 and 2,3,4 respectively. It is also possible to include more than one type of atom by using an array such as atoms = [0,1,3]. The index 5 would correspond to the total contribution from all the atoms.
Usage:
pyprocar.bandsplot(code='vasp',dirname='bands',,elimit=[-5,5],kticks=[0,39,79,119,159],knames=['G','X','M','G','R'],cmap='jet', mode='parametric',atoms=[1])
NOTE:
The reason for this format which includes the “total” atomic contribution as the last atom index is because the PROCAR file from VASP which PyProcar was originally based on had a “tot” row for each k-point/band block that summed up all the atom contributions. So the parser was meant to incorporate that as well. The other DFT parsers followed suit to be consistent with this format.
4. Orbital projection#
The projection of atomic orbitals onto bands is also useful to identify the contribution of orbitals to bands. For instance, to identify correlated orbitals is as follows (this is the same order from the PROCAR file). Quantum Espresso, VASP and Abinit follows this order.

In Elk, the
Usage: To project all five
pyprocar.bandsplot(code='vasp',dirname='bands',mode='parametric',elimit=[-5,5],kticks=[0,39,79,119,159],knames=['G','X','M','G','R'],cmap='jet',orbitals=[4,5,6,7,8])
One or many of the above can be combined together to allow the user to probe into more specific queries such as a collinear spin projection of a certain orbital of a certain atom.
Different modes of band structures are useful for obtaining information for different cases. The four modes available within PyProcar are plain, scatter, parametric and atomic. The plain bands contain no projection information. The scatter mode creates a scatter plot of points. The parametric mode interpolates between points to create bands which are also projectable. Finally, the atomic mode is useful to plot energy levels for atoms. To set maximum and minimum projections for color map, one could use vmin and vmax tags.
Export plot as a matplotlib.pyplot object#
PyProcar allows the plot to be exported as a matplotlib.pyplot object. This allows for further processing of the plot through options available in matplotlib. Usage:
ebs_plot = pyprocar.bandsplot(code='vasp',dirname='bands', mode='plain', show=False)
ebs_plot.ax.set_title('Using matplotlib options')
ebs_plot.fig.show()
Converting
PyProcar defaults to plotting using the reduced coordinates of the kdirect=False. However, an OUTCAR must be supplied for this case to retrieve the reciprocal lattice vectors to transform the coordinates from reduced to cartesian. Note that for the case of Elk, the output is automatically retrieved so it is not necessary to provide it for the conversion.
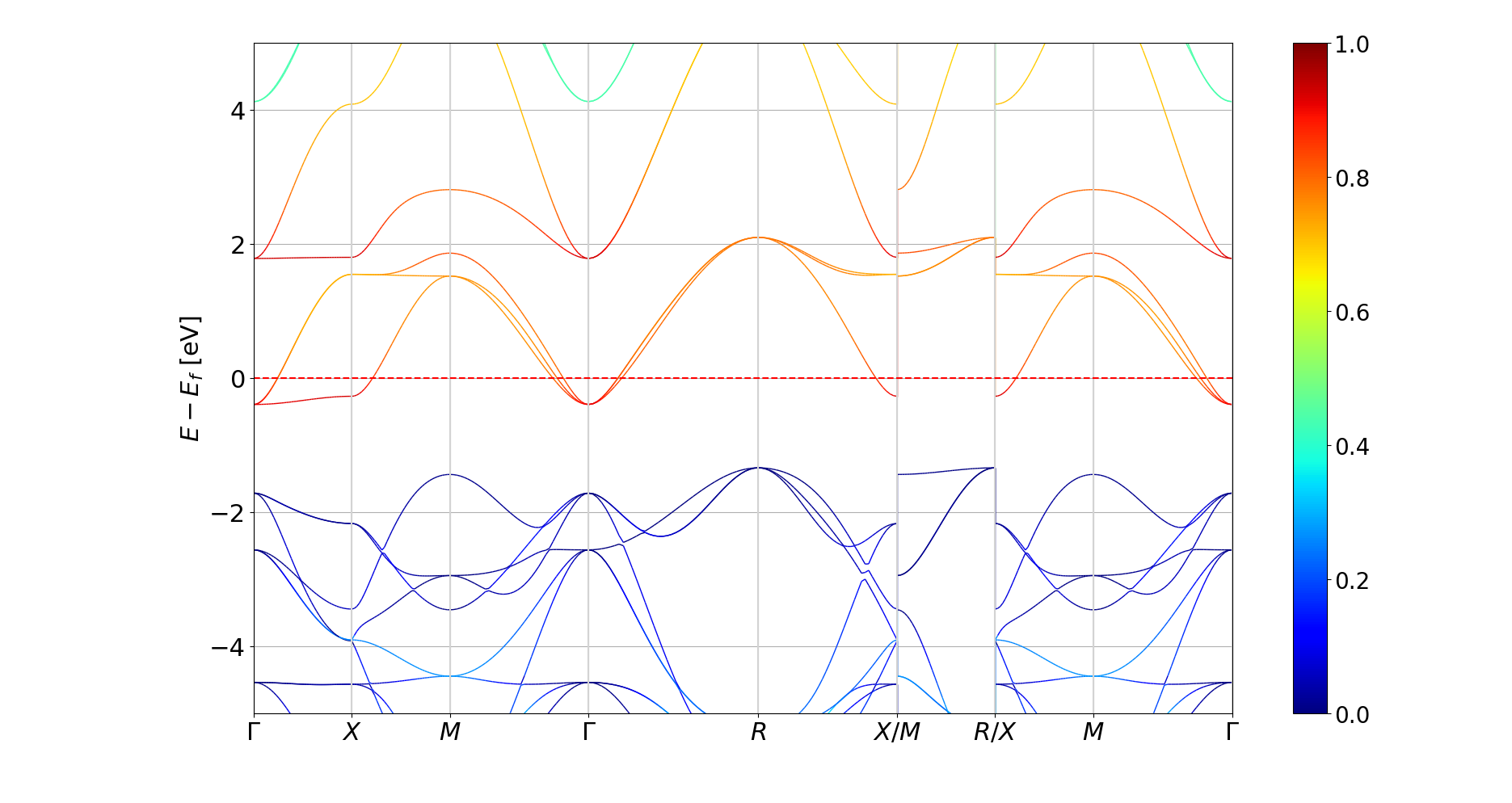
Plotting band structures with a discontinuous
PyProcar allows the plotting of band structures with a discontinuous KPOINTS file with the discontinuities variable. For example, the G-X-M-G-R-X M-R X-M-G with 40 grid points should be set as:
knames = ['$\\Gamma$', '$X$', '$M$', '$\\Gamma$', '$R$', '$X/M$', '$R/X$', '$M$', '$\\Gamma$']
kticks = [0, 39, 79, 119, 159, 199, 239, 279, 319]
discontinuities = [199, 239]
- pyprocar.scripts.scriptBandsplot.bandsplot(code: str, dirname: str, mode: str = 'plain', spins: List[int] | None = None, atoms: List[int] | None = None, orbitals: List[int] | None = None, items: dict = {}, fermi: float | None = None, fermi_shift: float = 0, interpolation_factor: int = 1, interpolation_type: str = 'cubic', projection_mask: ndarray | None = None, kticks=None, knames=None, kdirect: bool = True, elimit: List[float] | None = None, ax: Axes | None = None, show: bool = True, savefig: str | None = None, print_plot_opts: bool = False, export_data_file: str | None = None, export_append_mode: bool = True, **kwargs)[source]#
A function to plot the band structutre
- Parameters:
code (str, optional) – String to of the code used, by default “vasp”
dirname (str, optional) – The directory name of the calculation, by default None
mode (str, optional) – Sting for the mode of the calculation, by default “plain”
spins (List[int], optional) – A list of spins, by default None
atoms (List[int], optional) – A list of atoms, by default None
orbitals (List[int], optional) – A list of orbitals, by default None
items (dict, optional) – A dictionary where the keys are the atoms and the values a list of orbitals, by default {}
fermi (float, optional) – Float for the fermi energy, by default None. By default the fermi energy will be shifted by the fermi value that is found in the directory. For band structure calculations, due to convergence issues, this fermi energy might not be accurate. If so add the fermi energy from the self-consistent calculation.
fermi_shift (float, optional) – Float to shift the fermi energy, by default 0.
interpolation_factor (int, optional) – The interpolation_factor, by default 1
interpolation_type (str, optional) – The interpolation type, by default “cubic”
projection_mask (np.ndarray, optional) – A custom projection mask, by default None
kticks (_type_, optional) – A list of kticks, by default None
knames (_type_, optional) – A list of kanems, by default None
elimit (List[float], optional) – A list of floats to decide the energy window, by default None
ax (plt.Axes, optional) – A matplotlib axes, by default None
show (bool, optional) – Boolean if to show the plot, by default True
savefig (str, optional) – String to save the plot, by default None
export_data_file (str, optional) – The file name to export the data to. If not provided the data will not be exported.
export_append_mode (bool, optional) – Boolean to append the mode to the file name. If not provided the data will be overwritten.
print_plot_opts (bool, optional) – Boolean to print the plotting options