Note
Go to the end to download the full example code.
Plotting Atomic Levels#
This example illustrates how to plot atomic energy levels for the compound hBN-CNCN at the GAMMA point using the pyprocar package.
Preparation#
Before diving into plotting, we need to download the example files. Use the following code to do this. Once downloaded, specify the data_dir to point to the location of the downloaded data.
Downloading example#
import pyprocar
data_dir = pyprocar.download_example(
save_dir='',
material='hBN-CNN',
code='vasp',
spin_calc_type='spin-polarized-colinear',
calc_type='gamma'
)
Setting up the environment#
First, we will import the necessary libraries and set up our data directory path.
import os
import pyprocar
# Define the directory containing the example data
data_dir = os.path.join(pyprocar.utils.DATA_DIR, "examples", "hBN-CNCN")
Plotting in Atomic Mode#
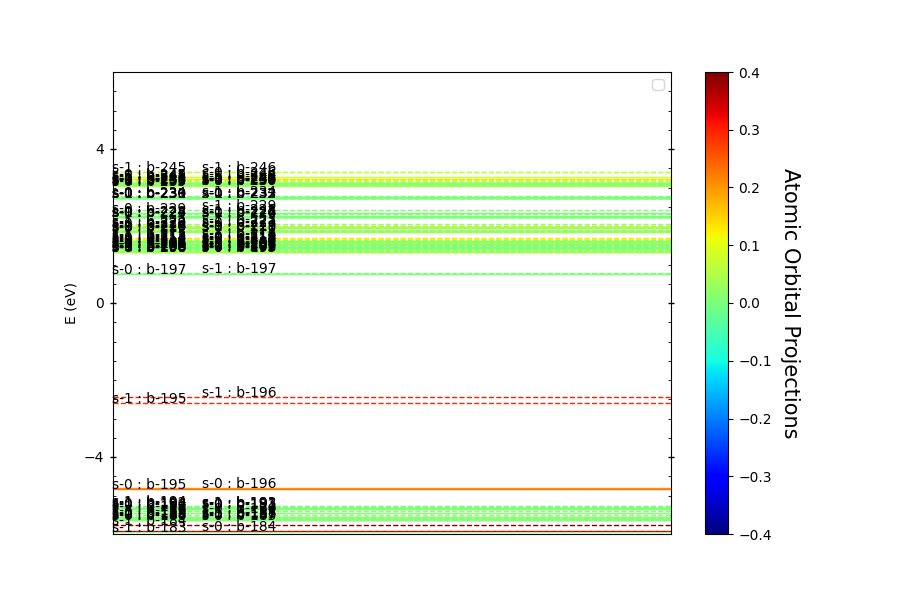
The atomic mode is used to plot the energy levels at a specific k-point. In this example, we focus on the GAMMA point. The plot will display energy levels for specific atoms in the range of -6 to 6 eV.
# Define the atom indices we're interested in
atoms = [96, 97, 0, 1, 2, 3, 42, 44]
# Plot the atomic bands
pyprocar.bandsplot(
code="vasp", # Specify the DFT code used. In this case, it's VASP.
dirname=data_dir,
mode="atomic",
elimit=[-6, 6], # Energy range for the plot
clim=[-0.4, 0.4], # Color limit for the plot
atoms=atoms, # Atoms for which we want to plot the energy levels
)
----------------------------------------------------------------------------------------------------------
There are additional plot options that are defined in the configuration file.
You can change these configurations by passing the keyword argument to the function.
To print a list of all plot options set `print_plot_opts=True`
Here is a list modes : plain , parametric , scatter , atomic , overlay , overlay_species , overlay_orbitals
----------------------------------------------------------------------------------------------------------
WARNING : `fermi` is not set! Set `fermi={value}`. The plot did not shift the bands by the Fermi energy.
----------------------------------------------------------------------------------------------------------
C:\Users\lllang\Desktop\Current_Projects\pyprocar\pyprocar\plotter\ebs_plot.py:701: UserWarning: Attempting to set identical low and high xlims makes transformation singular; automatically expanding.
self.ax.set_xlim(interval)
Atomic plot: bands.shape : (2, 246, 2)
Atomic plot: spd.shape : (2, 246, 98, 1, 9, 2)
Atomic plot: kpoints.shape: (2, 3)
(<Figure size 900x600 with 2 Axes>, <Axes: ylabel='E (eV)'>)
Total running time of the script: (0 minutes 1.129 seconds)